
Axon Regeneration
Understanding the mechanisms that regulate axon growth and axon-target interaction, towards the goal of rebuilding diseased or injured neural circuits
About us
Who We Are

We are a group of curious, kind, and respectful individuals who believe science is cool!
We believe in being open, honest, and strive to foster an environment where everyone thrives.
We believe happy people do better science.
What We Do
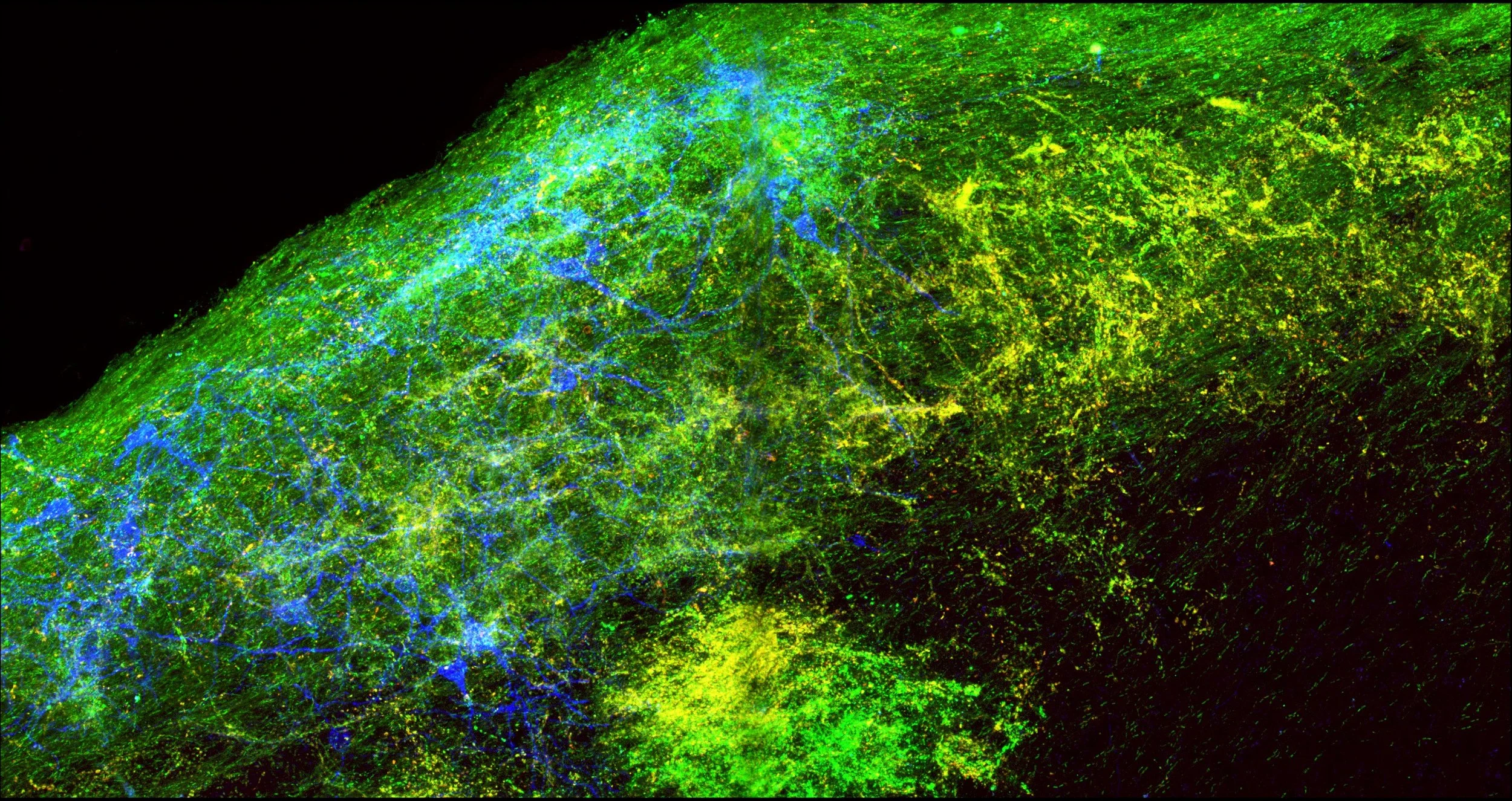
The Varadarajan Lab is interested in rebuilding neural circuits and restoring sensory function impaired by injury or disease. We use the eye-to-brain pathway to elucidate how damaged retinal ganglion cells in the eye can regrow their axons and restore connectivity in response to enhanced neuronal activity in the brain.
We are using the mouse visual system to tackle these concepts because of its beautiful, accessible, eye-to-brain pathway where retinal ganglion cell (RGC) axons in the eye project over long distances to reach their postsynaptic target neurons in the brain. Taking advantage of the molecular, genetic, viral, anatomical, and behavioral tools available to study the visual system, we aim to identify therapeutic strategies that can treat neurodegenerative diseases like Glaucoma, and other optic neuropathies that lead to blindness.
Why We Do It

Our objective is to identify therapeutic strategies that can be translated to restore vision in patients suffering from blinding diseases or eye injuries. Our work also has important implications for other neurodegenerative conditions in the brain and spinal cord.
We believe that to truly rebuild a damaged central nervous system, scientists from all walks of life have to work together to carefully reconstruct an intricate circuit piecemeal. This includes basic scientists with expertise in development, stem cells, computation, engineering, as well as clinicians.
We would love to work with people from different fields, share what we know, and learn what you know.